THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
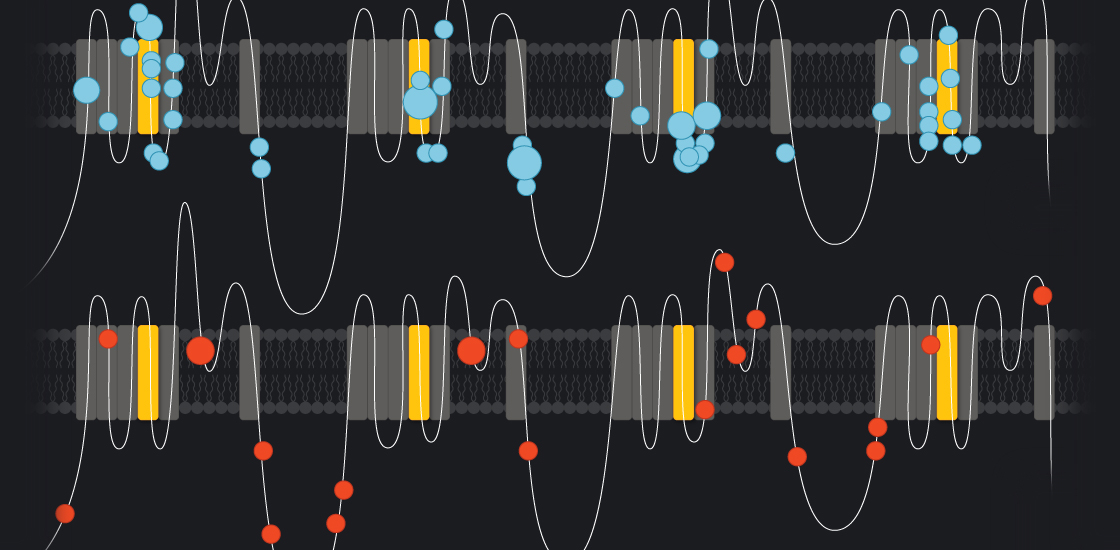
Some mutations in a gene called SCN2A appear to dampen brain activity and are linked to autism; others have the opposite effect and may lead to seizures during infancy1.
The findings, published 26 January in Biological Psychiatry, could guide treatment decisions for children with mutations in the gene. Knowing the effects of each mutation would help scientists identify which condition a child is at risk for, and could even help them predict the condition’s severity, says lead researcher Stephan Sanders, assistant professor of psychiatry at the University of California, San Francisco.
“Given a mutation and no other knowledge, I would be fairly confident in predicting infantile seizures versus developmental delay or autism,” Sanders says.
Understanding how an SCN2A mutation leads to autism may also reveal clues to the condition. Unlike many other autism genes, SCN2A has a straightforward function in the cell: It codes for a channel that allows the passage of sodium ions across the neuronal membrane. The ensuing rush of sodium into the cell helps propagate the electrical signals that neurons use to communicate.
Because the gene’s cellular function is so well understood, Sanders likens it to a “Rosetta Stone” for autism. “It gives us a window into what autism is,” he says.
Not all equal:
Two sequencing studies published in 2014 flagged SCN2A as a top autism gene. Researchers found harmful mutations in the gene in 12 individuals with autism, but not in their unaffected family members. Studies have also flagged SCN2A mutations in at least 15 more people with autism.
The only other gene with as strong an autism connection is CHD8. As it turns out, CHD8 regulates the expression of SCN2A and hundreds of other genes2.
In the new study, Sanders’ team mapped the location of 68 SCN2A mutations identified in people with a rare form of epilepsy called infantile seizures, and the location of 23 mutations seen in people with autism. They found that the mutations tied to autism cluster in different regions of the channel than do those associated with epilepsy. This suggests that the two sets of mutations have distinct effects.
The researchers expressed 12 autism mutations in kidney cells, which don’t ordinarily make the channel. They found that nine mutations produce channels that don’t transmit sodium ions; the other three diminish the channel’s function. (The kidney test may also be able to predict severity by giving a simple readout of the channel’s function.)
To predict the consequences in neurons, the researchers turned to a computational model. They fed the model data from the kidney cells. They also separately put in published information about SCN2A mutations tied to epilepsy.
The model determined that each of the autism mutations makes it more difficult for neurons to transmit currents. The epilepsy mutations, by contrast, make neurons overactive.
The study shows how important it is to look at a mutation’s effect on the cell, says Guoping Feng, professor of brain and cognitive sciences at the Massachusetts Institute of Technology, who was not involved in the study. “Now there is sufficient evidence that not all mutations are equal, even if they are in the same gene,” Feng says.
Sorting syndromes:
The finding that epilepsy mutations increase neuronal activity — which is known to cause seizures — makes sense clinically. The epilepsy mutations in SCN2A are all associated with infantile epilepsy.
None of the individuals with autism-linked mutations in SCN2A had seizures before age 1. Of the 19 people with autism for whom the researchers have clinical data, 4 had seizures later in life. It would be interesting to see whether other such individuals have SCN2A mutations with a distinct profile, says Geoffrey Pitt, professor of medicine at Weill Cornell Medicine in New York, who was not involved in the study.
The mutations’ link to early-onset seizures jibes with the timing of SCN2A’s expression. It is primarily expressed in excitatory neurons in infancy, but is replaced by other channels around age 1.
In autism, the story is less straightforward. Because autism mutations inactivate the channel, the brains of people with autism who have these mutations may be less excitable early in life. What happens later in life is uncertain, but one possibility is that in infancy the brain compensates for the effects by also decreasing inhibition. Later on, when SCN2A expression is replaced by functional channels, the under-inhibition may remain, leaving neurons’ excitatory activity to run unchecked.
This idea is consistent with theories that autism stems from an overexcitable brain, says co-lead researcher Kevin Bender, assistant professor of neurology at the University of California, San Francisco.
Ion involvement:
SCN2A is the not the only sodium channel linked to autism. Children with mutations in SCN1A, a sodium channel that works in neurons that dampen brain activity, have a severe form of epilepsy called Dravet syndrome and, sometimes, autism.
In a study published in December, researchers found a mutation in another sodium channel gene, called SCN9A, in one individual with autism. On follow up, the researchers discovered 29 mutations in the gene in 975 individuals with autism and only 2 in 1,119 controls3.
“This is solidifying the idea that sodium channels are involved in autism, and telling us we ought to be looking at this as seriously as we can,” says lead researcher William Catterall, professor of pharmacology at the University of Washington in Seattle.
Some teams are trying to recruit and characterize people with SCN2A mutations. The mutations seen in the individuals recruited so far match the types described in the new study, says Raphael Bernier, associate professor of psychiatry at the University of Washington. The individuals with autism in this study also did not have seizures before age 1.
By joining the discussion, you agree to our privacy policy.