THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
Immune cells called microglia may respond to neuronal damage from loss of the Rett syndrome gene, MeCP21. But contrary to some previous reports, they do not play a central role in initiating the syndrome, a new study suggests.
“Microglia may not be the underlying cause of the disorder or contributing in a major way,” says lead investigator Dorothy Schafer, assistant professor of neurobiology at the University of Massachusetts Medical School in Worcester.
Rett syndrome stems from mutations in MeCP2. Previous studies have hinted that microglia contribute to its features, which include breathing and movement problems, intellectual disability and autism-like behaviors.
For example, a 2012 study suggested that bone marrow transplants ease Rett-like features in mice missing MeCP2, by replenishing microglia. But four other research teams were unable to reproduce this result.
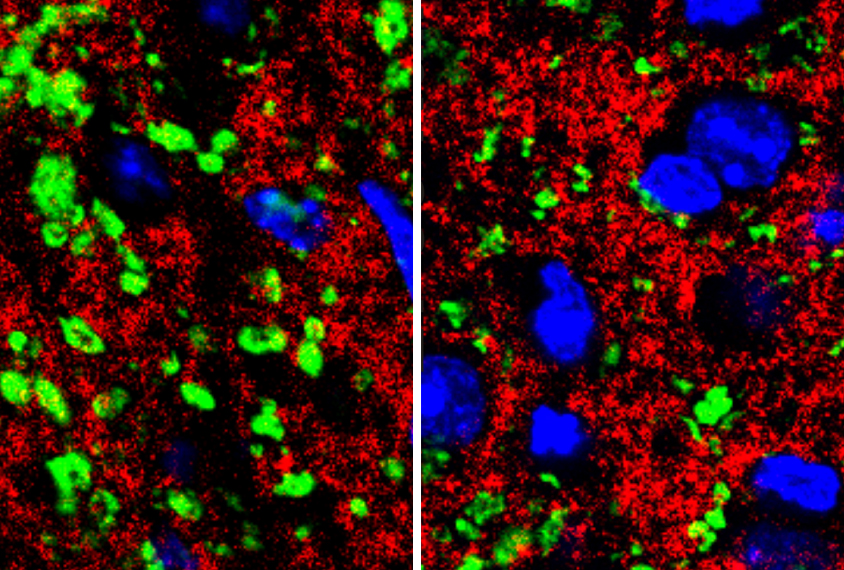
Schafer and her team set out to explore microglia’s role in Rett syndrome. They found that microglia in mice engulf too many connections between neurons only after the mice show symptoms. The findings appeared 26 July in eLife.
The cells may exacerbate damage in the Rett brain, however. Mice missing MeCP2 die prematurely, when they are about 60 days old — around the time the microglia become hyperactive.
“Our data do suggest that [microglia] could be contributing to late stages,” Schafer says.
Neural groomers:
Schafer and her colleagues discovered in 2012 that microglia trim excess synapses (neuronal junctions) in the visual systems of 5-day-old mice. The pruning pattern is selective: Microglia eliminate weak synapses rather than highly active ones2. This pruning may streamline the developing nervous system.
Two years later, another team reported that 30-day-old mice missing MeCP2 have weak synapses in the visual system3.
“That got us thinking, could microglia be preferentially attracted to those weakened synapses [in Rett syndrome]?” Schafer says.
In the new study, Schafer and her colleagues looked at mice missing MeCP2 in all of their cells. The researchers visualized microglia by labeling them with a protein that causes these cells to glow green. They also injected a red fluorescent dye into the animals’ eyes to stain the neurons that send signals from the retina to the brain. This allowed the researchers to see interactions between visual neurons and the surrounding microglia.
They saw no differences between Rett mice and controls in the pruning of synapses until the mice were about 56 days old. At that age, the Rett mice have more active microglia and fewer synapses than controls do, suggesting that the cells may accelerate the syndrome in its late stages, Schafer says.
First responders:
The researchers then studied Rett mice in which they could reinstate MeCP2 expression in microglia in response to the drug tamoxifen. This step has no effect on pruning and does not prevent any Rett-like features. The finding suggests that Rett syndrome stems from loss of MeCP2 in cell types other than microglia.
The researchers made mice in which tamoxifen shuts down production of MeCP2 only in microglia. These animals show no Rett-like features, and their microglia behave normally. This result further supports the notion that microglia dismantle synapses weakened by loss of MeCP2 in neurons.
“Microglia are properly responding to the abnormal loss of synapses typically driven by loss of the protein in neurons,” says Huda Zoghbi, professor of molecular and human genetics at Baylor College of Medicine in Houston, Texas, who was not involved in the study.
Schafer and her team aim to explore how microglia regulate synapses in other regions of the brain, and to determine whether these functions are disrupted in mouse models of Rett syndrome.
By joining the discussion, you agree to our privacy policy.