Robert S. McNeil / Science Source
THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A pair of existing drugs normalizes the appearance and activity of neurons derived from the skin of individuals with Rett syndrome. Researchers presented the unpublished findings yesterday at the 2016 American Society of Human Genetics Annual Meeting in Vancouver, Canada.
Rett syndrome is a rare form of intellectual disability that shares some features with autism. The most common form of the condition stems from mutations in the gene MeCP2. Disruptions to a second gene, called FOXG1, can produce an atypical form of Rett syndrome in girls and boys.
The new work suggests that mutations in either gene have similar effects on neuronal function. The researchers were able to reverse these effects in a stem cell model of Rett using drugs approved for other conditions, such as cancer.
“We found common pathways disrupted by MeCP2 and FOXG1, and these targets are amenable to treatment,” says Elisa L’Anducci, who presented the findings. L’Anducci is a graduate student in the laboratory of Alessandra Renieri, director of the medical genetics unit at the University of Siena in Italy.
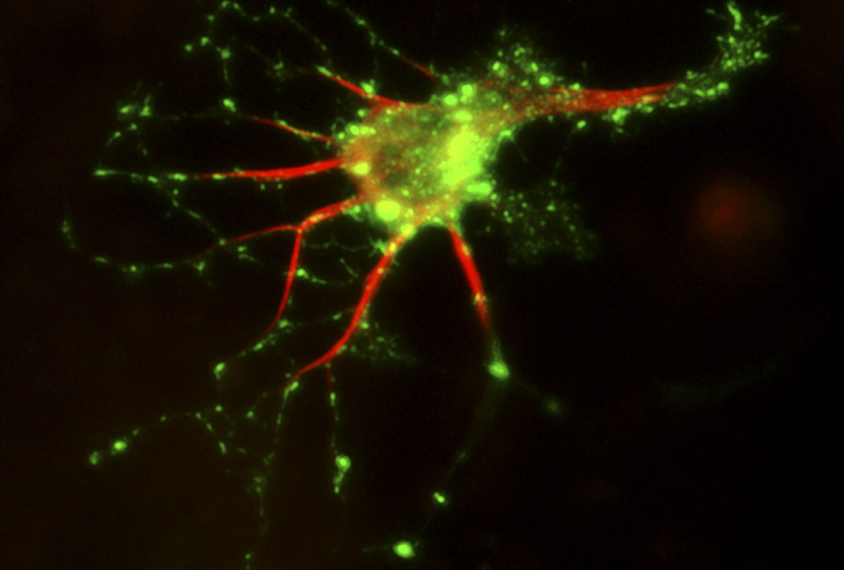
L’Anducci and her colleagues obtained skin cells from two people with missense mutations in MeCP2, two people with shortened versions of FOXG1 and two controls. The team coaxed these cells to form induced pluripotent stem (iPS) cells and then transformed the stem cells into neurons.
Compared with control neurons, those carrying MeCP2 or FOXG1 mutations form unusually short projections, called axons, and a weak extracellular matrix — the supportive mesh between cells. The mutant cells also form disorganized networks with other cells.
The researchers then measured gene expression in the three types of neurons. They identified 127 genes that are ramped up in cells carrying MeCP2 or FOXG1 mutations and 247 genes that are tamped down in the mutant cells.
Skeleton key:
Many of the gene expression changes are likely to boost the activity of gamma-aminobutyric acid (GABA), a chemical messenger that quiets brain activity. Some of the genes control the production of GABA, and others encode GABA receptors or transporters. Several studies have linked GABA signaling to Rett syndrome and autism.
Other gene expression changes seen in the mutant cells would prevent the assembly of microtubules, which form the cells’ supportive skeleton and interact with the extracellular matrix. Microtubules also transport signaling molecules to neuronal junctions, or synapses, and are part of signal-receiving structures called dendrites.
The researchers sifted through a library of experimental and approved drugs for any that might normalize GABA signaling or microtubule assembly. They landed on flumazenil, which acts on the GABA receptor to relieve sedation, and the cancer therapy panobinostat, which stabilizes the cell’s skeleton.
The researchers treated the neurons with either drug and evaluated the cells’ appearance, connections and electrical activity. Their preliminary findings suggest that the drugs counteract the effects of these mutations. They plan to test the drugs’ effects on behavior in mice with mutations in either Rett gene.
For more reports from the 2016 American Society of Human Genetics Annual Meeting, please click here.
By joining the discussion, you agree to our privacy policy.