THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
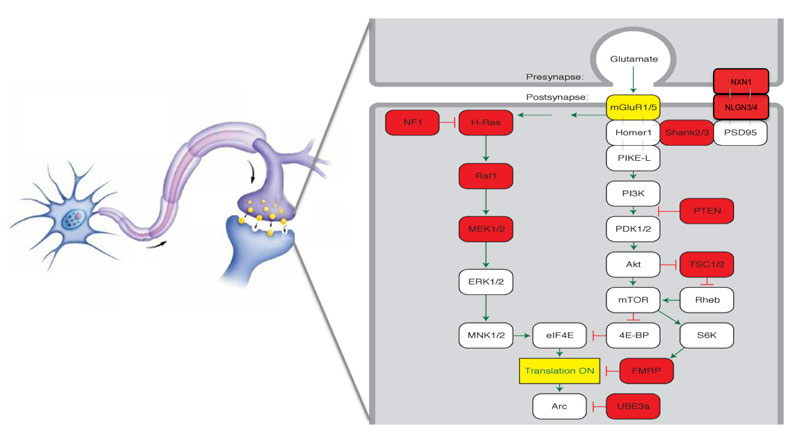
Some forms of autism are caused by too many proteins at the synapse, the junction between neurons, whereas other forms result from too few, according to a study published 23 November in Nature1.
The findings suggest that drugs that effectively treat people with one form of autism may not help, and may even harm, individuals with another form, the researchers say.
“One implication is, boy, it’s going to be important to know where you are on this spectrum to devise the right therapy,” says lead investigator Mark Bear, professor of neuroscience at the Massachusetts Institute of Technology.
The work, first presented at the 2010 Society for Neuroscience annual meeting in San Diego, focuses on mouse models of two genetic diseases: fragile X syndrome, the most common inherited cause of autism, and tuberous sclerosis complex (TSC), characterized by benign tumors, seizures and, often, autism.
Autism is notoriously diverse. But the new study suggests that many forms of the disorder stem from a spectrum of disruptions to the same biological pathway, Bear says. “The hope is we’re gaining insights that will be broadly applicable.”
Other experts are also optimistic that these findings could begin to unravel common threads underlying different types of autism.
“This is a very exciting and unexpected finding, and has really important implications for therapies in different genetic disorders associated with autism,” says Mustafa Sahin, assistant professor of neurology at Children’s Hospital Boston, who was not involved in the study.
Opposites attract:
In 2002, Bear’s group showed that, compared with controls, mouse models of fragile X syndrome have too many proteins at the synapse. Specifically, the mutants show excessive synthesis of proteins controlled by the mGluR5 receptor, which uses the neurotransmitter glutamate for signaling2.
Five years later, the researchers reversed the learning problems in these mice with drugs that dampen mGluR5 activity. Several clinical trials are testing similar drugs on children with the syndrome.
In that same time period, other teams showed that a drug called rapamycin reverses the learning deficits in mouse models of TSC. Rapamycin inhibits a protein called mTOR that is important for cell growth and protein synthesis. This led to the popular hypothesis that TSC, like fragile X syndrome, is caused by excessive protein synthesis at the synapse3.
In 2009, Bear applied for a federal stimulus grant to compare protein synthesis in fragile X and TSC mice. The proposal was rejected. “The reviewers thought we were being too conservative, because [they thought] of course TSC would be just like fragile X,” Bear says.
On the contrary, the new study shows that TSC mice have much less mGluR5-regulated protein synthesis at the synapse than controls do. In this way, TSC is the opposite of fragile X, Bear says.
“This paper gives rise to a lot of questions. And it shows that maybe some things we thought we understood well, we don’t understand so well,” says Bernardo Sabatini, professor of neurobiology at Harvard Medical School, who was not involved with the study.
TSC is caused by mutations in one of two genes: TSC1 or TSC2. Mice missing either gene have significant memory problems. In June, Sabatini’s group reported that mice lacking TSC1 in neurons in the hippocampus, a region important for learning and memory, show changes at the synapse that are similar to those reported in Bear’s study4.
In the new study, Bear’s team analyzed male mice missing one copy of TSC2. In hippocampal slices, the researchers showed that mGluR5-controlled protein synthesis is significantly lower in these mutant mice than in controls.
It’s unclear why the mutants make fewer proteins, but the effect seems to be dependent on mTOR. The researchers showed that when the mice are given rapamycin, the mTOR inhibitor, the level of protein synthesis normalizes.
It could be that this effect is specific to the hippocampus, Sahin notes. “The authors are looking at one mouse model of TSC using a very specific region of the brain under certain conditions,” he says. “I’m not sure how generalizable that is yet.”
Bedside to bench:
Perhaps most importantly, Bear’s team demonstrated that the protein synthesis and learning problems in TSC2 mice can be normalized in two ways, one genetic and the other pharmacological.
In the genetic approach, the researchers knocked out the fragile X gene FMR1 from the TSC2 mutants. The lack of FMR1 typically leads to too much protein synthesis, and the TSC2 loss too little, so these effects are effectively canceled out in the double mutants. The learning abilities of these mice are no different than those of control mice, the study found.
“This is fascinating because it suggests that maybe in humans, we can have counterbalancing mutations, where one may be actually protective against another,” Sabatini says.
Bear’s team was also able to alleviate the learning problems in TSC2 mice with a type of drug dubbed PAM, or ‘positive allosteric modulator,’ that stimulates mGluR5 receptors.
Many drug companies are working on mGluR5 PAM drugs, according to Bear. If these compounds meet safety requirements, he says, they could be used instead of mTOR inhibitors to treat TSC.
Sahin is leading a clinical trial of everolimus, an mTOR inhibitor that is similar to rapamycin, in children and adults with TSC. Unfortunately, this drug, like rapamycin, suppresses the immune system.
“mTOR inhibitors have been the mainstay of treatment for mouse models of TSC and they work remarkably well. However, they do have some side effects,” Sahin says. “So it would be ideal to use a group of drugs with fewer side effects.”
These drugs might also be effective in children who have autism unrelated to fragile X or TSC. But for now, there’s no clinical or biological marker that could help researchers determine which of these cases are similar to TSC and which are more like fragile X.
“That’s the big problem,” Sabatini says. “It’s kind of terrifying — both because you don’t know, in the idiopathic case, and secondly, because it suggests that if you treat a little bit too much, if you get the dosage a little bit off, you’re going to mess the kids up again.”
Clinicians are likely to be facing these issues soon. If drugs that act on the mGluR5 pathway are approved for treating fragile X syndrome, doctors may prescribe them to children with other forms of autism, and researchers may learn more about which subgroups of children respond to the drugs.
“We often talk about going from bench to bedside,” Bear says, referring to treatments that are based on mechanisms discovered in the lab. “In this case, we have the opportunity to go from bedside to bench.”
Correction: This article has been modified from the original. It has been changed to correct an error about mice lacking TSC1 neurons in the hippocampus (ref. 4).
References:
1: Auerbach B.D. et al. Nature Epub ahead of print (2011) Abstract
2: Huber K.M. et al. Proc. Natl. Acad. Sci. U. S. A. 99, 7746-7750 (2002) PubMed
3: Hoeffer C.A. and E. Klann Trends Neurosci. 33, 67-75 (2010) PubMed
4: Bateup H.S. et al. J. Neurosci. 31, 8862-8869 (2011) PubMed
By joining the discussion, you agree to our privacy policy.