Editor's Note
This article was originally published in July 2022, based on a preprint. We have updated the article following publication of the study in Neuron in July 2023.
An experimental drug improves sensory sensitivities in fragile X model mice — but only if it’s administered after certain brain circuits have formed, according to a new unpublished study.
An experimental drug boosts the activity of neurons that are underdeveloped in fragile X syndrome and reverses sensory sensitivities in a mouse model of the condition — but only if it’s given after certain circuits have formed, according to a new study. The findings reveal a potential window for treating fragile X traits and identify a compound that may be relevant to sensory issues in autism, the researchers say.
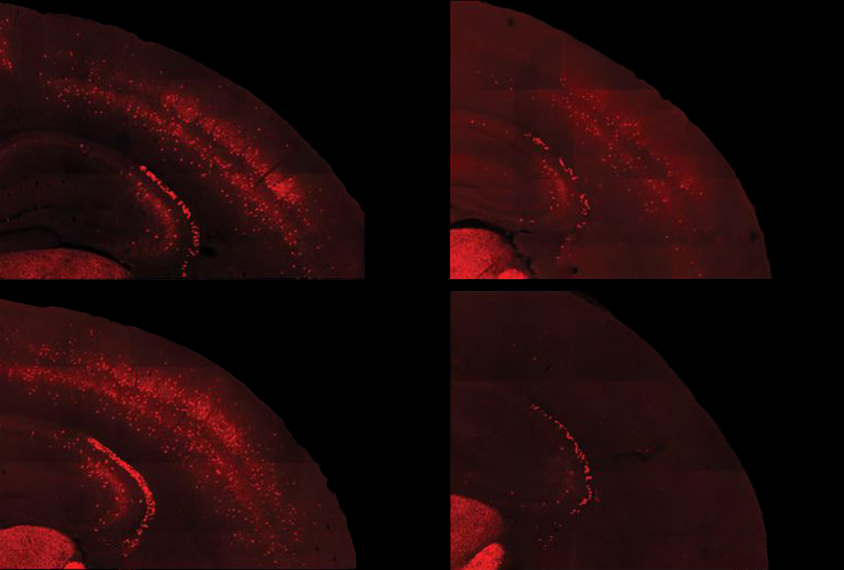
People with fragile X syndrome — an autism-linked condition caused by mutations in the FMR1 gene — typically have intellectual disability and sensory sensitivities. These sensitivities may stem from underactive parvalbumin (PV) interneurons, a type of inhibitory neuron, according to studies of mice missing the FMR1 gene. Those neurons also mature unusually slowly in fragile X model mice, a 2017 study found. And some research suggests that autistic people have too few PV interneurons throughout their cortex, though those results have been contested.
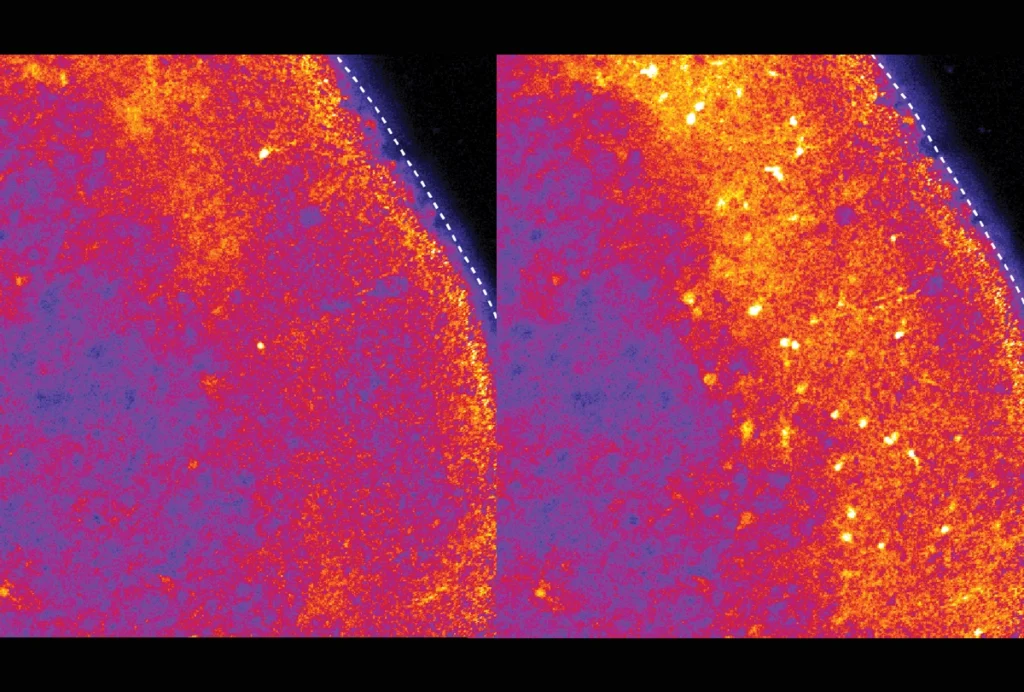
In the new study, the researchers confirmed that fragile X model mice have both underactive PV neurons and not enough of them, and they tied those changes to an excess of PV interneurons dying in the second week of life, shortly after they migrate to the cerebral cortex. Correcting the underactivity of PV precursor cells in 5- to 9-day-old mice prevented the cells from later dying off en masse, the researchers found, but it did not improve the animals’ sensory responses.
“We were very puzzled by this,” says lead investigator Carlos Portera-Cailliau, professor of neurology and neurobiology at the David Geffen School of Medicine at the University of California, Los Angeles.
In contrast, treating the animals slightly later in life — between the ages of 15 and 20 days old, once the PV cells begin forming connections with other neurons — did not prevent cell death, but it did reverse the animals’ atypical sensory responses, the team found. They posted their findings to a preprint server in May.
Some questions remain, says Renata Batista-Brito, assistant professor of neuroscience and genetics at Albert Einstein College of Medicine in New York City, who was not involved in the work. For one, she says, the cells that develop into PV neurons also give rise to another type of interneuron, and it’s unclear if their activity is being modulated as well. But as a first investigation, the work is “really exciting,” Batista-Brito says. (That type of interneuron [somatostatin] did not show reduced activity in the knockout mice, wrote Portera-Cailliau in an email to Spectrum after the study published in July 2023 in Neuron.)
T
Fragile X model mice, however, do not have the same adaptive response, previous research from Portera-Cailliau’s lab has shown. “They don’t reduce their firing gradually to tune out stimuli,” says study investigator Nazim Kourdougli, a postdoctoral researcher in the lab.
That lack of adaptation stems from a disconnect between the inhibitory PV neurons and the excitatory neurons they form a circuit with, the new work shows. Optogenetically stimulating PV cells in the somatosensory cortex of 10-day-old wild-type mice led to a decrease in the activity of excitatory cells in the region. But in fragile X model mice, activating PV cells had no effect on excitatory neurons, which may indicate delayed development of the somatosensory circuit.
A drug that boosted the activity of PV precursor cells in 5- to 9-day old mice induced a more typical pattern of gene expression in the animals’ cortex, the researchers found. The treatment also prevented the animals from losing PV cells, but it did not repair the somatosensory circuit or improve the animals’ adaptive response to a whisker touch.
“Even though we could restore [the PV cells’] density, that was not enough to help the circuit,” Portera-Cailliau says — possibly because the circuit had still not yet fully formed.
But boosting the inhibitory cells’ activity in 15- to 20-day old mice using a different molecular compound, AG00563 — which activates a type of potassium channel that is essential for the function of PV interneurons — had the opposite effect. After 5 days of treatment, the animals had more cells that responded to the stimulus and an increased adaptive response.
Portera-Cailliau and his colleagues suspect that even though the animals had already lost a large proportion of their PV cells, the remaining ones were still able to properly wire up with excitatory cells to shape the somatosensory circuit.
“I do not believe that we are compensating for the lack of PV density,” Kourdougli says. However, increasing the activity of PV cells in a strong and specific manner “appears to be enough to improve those deficits we studied.”
T
In a study published last month, Klann and his colleagues showed that a lack of FMRP, the protein encoded by FMR1, in PV interneurons contributes to other fragile-X-linked traits in mice, such as atypical social behaviors and anxiety-like behavior.
That work further “supports the notion that these interneurons could be therapeutic targets,” Portera-Cailliau says.
One caveat remains when it comes to PV cell loss in people with fragile X syndrome, says Gordon Fishell, professor of neurobiology at Harvard University, who was not involved in the work. “Loss of PV in human can mean loss of cells, or it could just be loss of PV [protein] itself,” he says, referring to the parvalbumin protein produced by the neurons that researchers use to identify the cells. The researchers acknowledge this in their paper discussion, he says, and they verified that, in fragile X model mice, the cells themselves are lost. But more research is needed to support the same claim in humans, he says.
It’s also unclear whether, based on these results, future treatments for fragile X syndrome should prioritize preventing the loss of PV cells, Fishell says. Boosting PV cells’ activity in 15-day-old mice led to restored circuits and improved sensory responses, even though the bulk of PV cells had presumably died, he says, suggesting that targeting circuit function is more important for improving sensory traits. “But let’s be honest — they’re looking at whisking. They’re not looking at cognitive engagement.”
Preventing the death of PV cells in fragile X syndrome may ultimately prove to be an important strategy for long-term treatment of the condition, Kourdougli says. But because surviving cells will still have a lack of FMRP, they will still function immaturely.
As a result, he says, his group’s later approach to intervention may end up being a good strategy. “Alternatively, further studies will need to be performed to identify the underlying mechanisms of this immaturity and target those.”