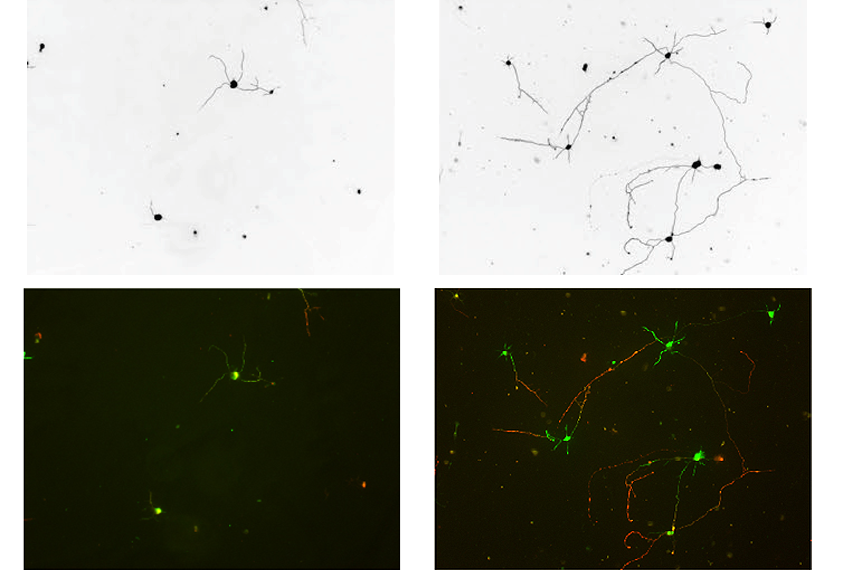
Neurons garner much of the attention in autism research, but they rely on star-shaped brain cells called astrocytes to grow properly and form synapses. Neurons from wildtype mice, for example, fail to thrive when exposed to astrocytes cultured from mice or people with autism-linked conditions, including Rett syndrome, fragile X syndrome and Down syndrome.
The explanation may lie with a protein the syndromic astrocytes secrete, according to a study published in August in Nature Neuroscience and led by Nicola Allen, associate professor of molecular neurobiology at the Salk Institute in La Jolla, California.
Astrocytes taken from mouse models of Rett, fragile X and Down syndromes collectively showed increased expression of 88 secreted proteins and decreased expression of 32 others, Allen’s team found. Notably, astrocytes from all three models secreted higher-than-normal amounts of IGFBP2, a protein that inhibits insulin-like growth factor (IGF).
An investigational drug that mimics the action of IGF, called trofinetide, already seems to ameliorate problems related to Rett syndrome in girls and women. Spectrum spoke with Allen about her results’ implications for future drug targets for Rett and other neurodevelopmental conditions.
This interview has been edited for length and clarity.
Spectrum: Why did you focus on these three syndromes?
Nicola Allen: We picked them because these were the three main syndromes for which other people had shown the astrocytes had kind of a similar effect. What we wanted to do was see if, across these syndromic forms that have very different genetic causes, there was any convergence on the level of how the astrocytes change. And if so, that would then give us insight in cases in which we don’t know the cause, but we know what the effect is, into ways we could think about trying to target that therapeutically.
S: Your study found little overlap between the astrocytes’ changes in gene expression and protein secretion. What does this tell us?
NA: I think it’s just reminding us that there are many steps between RNA production and the protein being made and modified and sent to the right place where it needs to act. This suggests that the regulation that’s happening isn’t just happening at the stage of the RNA being made, it’s happening further downstream. So when you’re thinking about cell-to-cell communication as we are, you want to analyze that final output step — in this case, the proteins being released from the astrocytes.
S: Among the proteins that were expressed differently, you homed in on IGFBP2. Why?
NA: The reason we picked IGFBP2 is because, of all the really important work that has been done, particularly with Rett, looking at IGF1 and IGF2 has shown that these pro-growth factors can have beneficial effects in rescuing some deficits. One of IGFBP2’s known roles is to act as an inhibitor of IGF. We were very interested in the idea that, in Rett and the other disorders, astrocytes could be releasing too much of this inhibitor, and this inhibitor then stops this IGF from working.
We showed that if we took proteins from the wildtype astrocytes and just added extra of this one protein, IGFBP2, then this media inhibited neuronal growth, telling us that this protein by itself had a pretty potent affect.
And then in the opposite way, we used an antibody that binds the IGFBP2 at the place where it would normally interact with IGF, stopping the binding protein from inhibiting the IGF. So we could show in the cell-culture experiment that this could then overcome some of the inhibitory effect in the Rett syndrome astrocytes.
S: Did this outcome look the same across all three conditions?
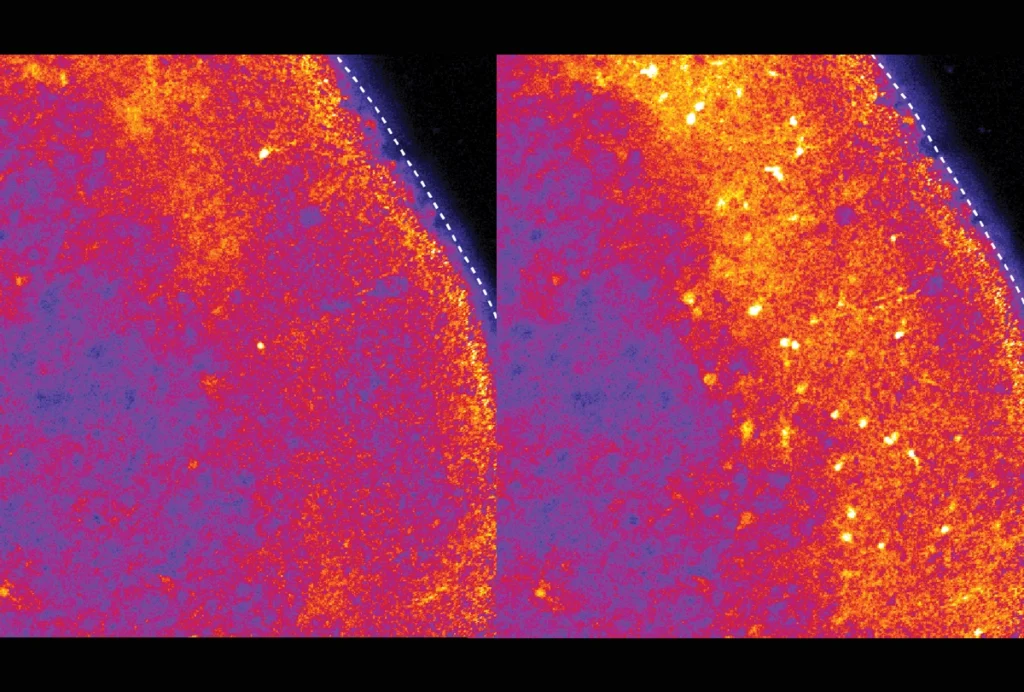
NA: The main effect seemed to be in the Rett astrocytes, actually. What was interesting — and this shows the importance of testing all the things that you identify in a screen — was that we saw the same thing in vivo, in the developing cortex; when we looked at IGFBP2 outside the astrocyte there was more in the Rett mice compared with the wildtype mice. And in the Down syndrome model, we saw a buildup of the protein inside the astrocytes; and in the fragile X, in the tissue, we didn’t see a significant change overall.