Listen to this story:
Over- or under-expression of UBE3A, a gene linked to autism and autism-related syndromes, alters synapse pruning at the signal-releasing ends of neurons, according to a study in fruit flies.
The findings add to mounting evidence that UBE3A plays an important role at the presynaptic side of synapses. Previous studies have found that flies and mice with mutated UBE3A have defects in the growth and function of signal-sending synapses.
“There’s enough clues out there to suggest that [UBE3A] is doing something really important in the presynaptic compartment,” says Jason Yi, assistant professor of neuroscience at Washington University in St. Louis, Missouri, who did not take part in this research.
But, he adds, many previous findings obtained in flies have not generalized to mice or people. “It remains to be seen whether that’s due to the [fly] system being different or if we just haven’t looked hard enough in mammalian systems.”
A
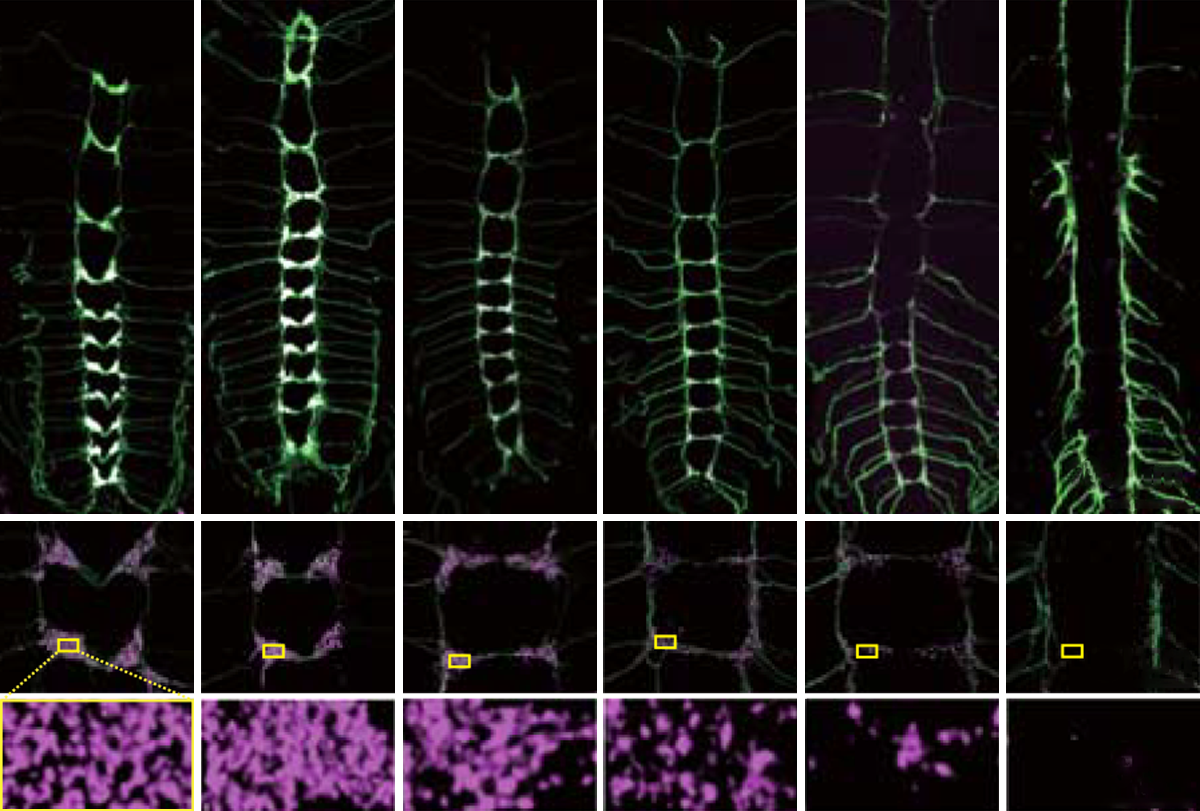
cross species, there is an overgrowth of synapses early during development, followed by the elimination of weak or unnecessary connections. Synaptic pruning is thought to be essential for brain function, and disruptions in this process have been linked to autism.In fruit flies, synaptic pruning occurs as larvae develop into adults. In the first 24 hours of metamorphosis, signal-sending synapses are removed in a class of sensory neurons called C4da neurons, which are responsible for sensing thermal, mechanical and light stimuli.
But flies that express low levels of UBE3A show pruning deficits, researchers led by Kazuo Emoto of the University of Tokyo in Japan reported in Science this month. (Emoto and his colleagues didn’t respond to requests for comment for this article.) In people, a mutated or missing maternal copy of UBE3A results in Angelman syndrome, which is characterized by developmental delay, motor difficulties, seizures and often autism.
UBE3A is typically transported by a motor protein from the cell bodies to the signal-sending ends of C4da neurons, the researchers found. But in flies with Angelman-linked UBE3A mutations, UBE3A transport and localization at the presynaptic side are reduced.
By contrast, expressing high levels of UBE3A in C4da neurons causes precocious elimination of existing synapses at the signal-sending ends of the cells, the team found. UBE3A is duplicated in people with dup15q syndrome, some of whom have autism, and mutations that enhance UBE3A activity are linked to autism.
Flies with increased UBE3A expression have reduced synaptic function and diminished sensitivity to heat. Some mouse models of autism also have reduced heat sensitivity, past work shows.
UBE3A belongs to a class of enzymes that tag proteins for destruction. So in a series of further experiments, the team homed in one of UBE3A’s putative targets: a protein called TKV.
Expressing high levels of TKV in the C4da neurons of wildtype flies caused defects in synapse elimination comparable to those observed in neurons with a disruptive UBE3A mutation. During early metamorphosis, TKV levels were reduced in wildtype flies but not in flies with a mutated copy of UBE3A. The findings suggest that UBE3A removes synapses by dampening the levels of TKV in neurons, the researchers write in their paper.
T
KV is a receptor for bone morphogenetic proteins (BMPs), a group of signaling molecules involved in brain development. Upregulation of the BMP signaling pathway has been observed in mouse models of Rett syndrome and fragile X syndrome, two autism-linked conditions.The link between UBE3A and the BMP signaling pathway is intriguing, says Ype Elgersma, professor of neuroscience at Erasmus University in Rotterdam, the Netherlands, who was not involved in the study. But, he cautions, the study provides little evidence that UBE3A targets TKV for degradation.
It’s also unclear whether the findings obtained in flies would translate to people, Yi says. The fly UBE3A protein is quite different from its human equivalent, he says, and in mammals only the UBE3A allele inherited from the mother is functional in neurons — a phenomenon known as imprinting. “But in flies there’s no imprinting, which implies that there’s some differences in the underlying biology and regulation of UBE3A between flies and humans.”
These species-specific quirks may also explain why the loss of UBE3A in flies results in decreased synaptic pruning, which leads to a surplus of synapses, whereas loss of UBE3A in mice and people typically causes a decrease in synapse number, Elgersma says. “If this phenotype is opposite in flies versus mice and human, that makes things complicated when you think about treatments,” he says.
Regardless, a better understanding of UBE3A’s role at the synapse is needed, says Lawrence Reiter, professor of neurology at the University of Tennessee Health Science Center in Memphis, who did not take part in the study. Scientists know that UBE3A is involved in strengthening or weakening neuronal connections, Reiter says. But, he adds, “we are not sure what it’s doing or which substrates are involved.”