THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A single gene accounts for most of the features seen in people missing a segment of chromosome 15 known as 15q13.3, two new studies suggest1,2.
People with this deletion often have intellectual disability, epilepsy, language impairment and, in about 15 percent of cases, autism.
Based on genetic sequences from more than 6,000 people with autism, one of the new studies points to a role for the gene, OTUD7A, in autism. Both studies report that OTUD7A is important for brain function in mice and is the key gene responsible for the unusual behaviors seen in mice missing 15q13.3.
The results contradict several earlier reports, which pointed to a different gene — CHRNA7 — as the culprit in the region.
“It came as quite a surprise to see these two papers,” says Catalina Betancur, director of research at INSERM in Paris, who was not involved in either study. “Both give a lot of evidence that [OTUD7A] plays an important role in the microdeletion syndrome.”
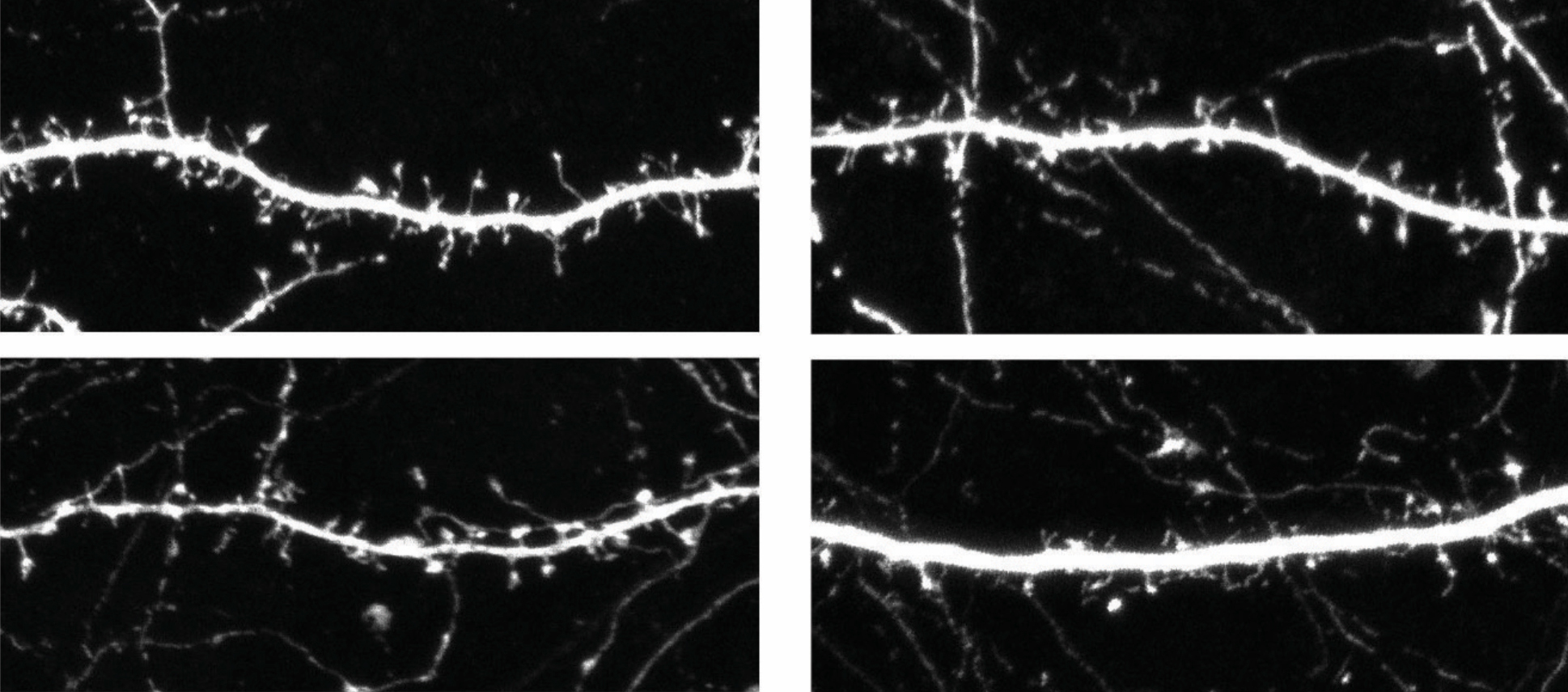
Loss of the gene stifles communication between neurons and decreases the number of dendritic spines — the thorny protrusions that connect neurons in the brain — the researchers found. Both studies appeared 1 February in the American Journal of Human Genetics.
“If we understand the biology better, it offers us better hope of potentially identifying a therapeutic intervention,” says David Miller, associate professor of pediatrics at Boston Children’s Hospital, who was not involved in either study.
Persistent relevance:
In the genetics study, researchers analyzed the genomes of 84 families that include two children with autism and two unaffected parents. They also looked at sequences of the exomes — the small fraction of the genome that encodes proteins — of 5,953 families that have only one child with autism and two unaffected parents.
The researchers spotted eight spontaneous, or de novo, mutations in the 15q13.3 region that appear in people with autism but not in their parents. Three of the mutations are in OTUD7A, and one of them is likely to disrupt the protein’s function.
The researchers did not find any OTUD7A mutations among the sequences of 250 control families or those of the 5,205 unaffected siblings of people with autism.
The team then used existing datasets to assess brain expression levels of seven genes in 15q13.3 and their relative vulnerability to mutation. They found higher levels of expression and a lower tolerance to mutation for OTUD7A than for the other genes.
An analysis of protein levels in postmortem tissue from three individuals revealed that OTUD7A is the only gene in 15q13.3 to encode a protein that works exclusively in the brain. It also belongs to a group of proteins regulated by FMRP, the protein missing in fragile X syndrome, and that is known to harbor autism mutations.
“OTUD7A was the only gene that kept popping out as being significant,” says Karun Singh, a principal investigator at McMaster University’s Stem Cell and Cancer Research Institute in Hamilton, Ontario, who co-led the study.
Branching out:
The same team also looked at mice missing a copy of the 15q13.3 region. They have fewer neuronal branches than controls, and their spines are immature, the team found. These branches and spines develop normally when mutant mouse embryos are given a working copy of human OTUD7A.
By contrast, two other genes in the region — KLF13 and FAN1 — do not rescue normal development. A functional copy of CHRNA7 improves neuronal branching but not spine development.
Taken together, the findings suggest that OTUD7A alone regulates the development of spines, but works with CHRNA7 to form neuronal branches, Singh says.
In the second study, researchers deleted one or both copies of OTUD7A in mice and analyzed the mice’s behaviors on 16 standard tests.
The mutant mice have many of the same features as people with a full 15q13.3 deletion, and mice missing both copies of the gene are more severely affected than those missing a single copy. For example, the mutant mice are underweight and are late to reach developmental milestones, including some motor skills. They also have seizures, a weak grip and trouble balancing on a rotating rod. And when separated from their mothers, the pups are less likely than controls to vocalize. People with 15q13.3 deletions also have seizures, motor difficulties and language problems.
Unlike people with the deletion, however, the mice have no learning and memory problems, social deficits or repetitive behaviors.
“The knockout mouse recapitulates some but not all of the clinical features we see in patients,” says lead investigator Christian Schaaf, professor of clinical genomics and medical director of human genetics at the University of Cologne in Germany.
Protein protector:
Schaaf and his colleagues also analyzed the brains of the mutant mice. Like mice missing a copy of the region, those lacking only OTUD7A also have fewer spines than controls do. However, they have an ordinary number of neuronal branches.
“This particular sub-segment of the neuron — the dendritic spines — is something that seems to be impacted in many disorders that relate to autism,” Schaaf says.
Schaaf’s team also found that neurons in the mutant mice transmit fewer excitatory signals than in controls.
OTUD7A encodes a protein that removes a tag called ubiquitin, which marks other proteins for destruction. It joins a growing list of autism-linked genes that function as part of the ubiquitin pathway, including MAGEL2 and UBE3A.
“More and more evidence is showing that [ubiquitin pathways] need to be very finely regulated,” Schaaf says. “Whenever you mess with that system, it can have an impact on the brain.”
Schaaf and Singh both say their next goal is to identify the proteins that OTUD7A regulates. This may yield clues to how OTUD7A affects spines, and point to therapeutic strategies.
By joining the discussion, you agree to our privacy policy.