THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
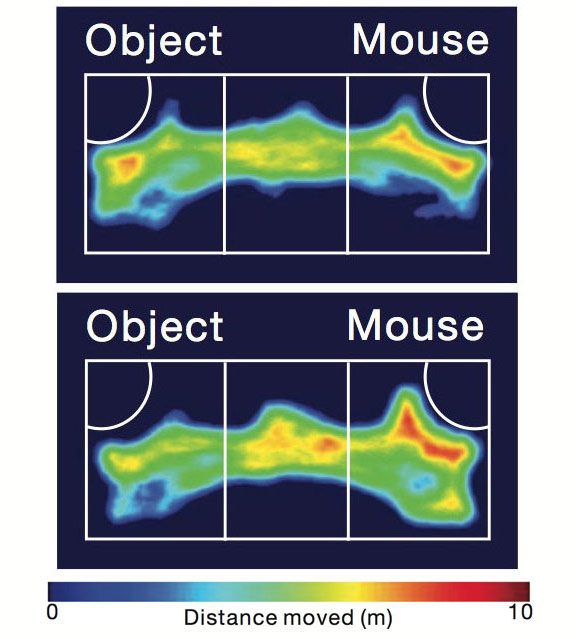
Social slackers: In a three-chamber test of social interactions, control mice (bottom) spend more time interacting with a new mouse than with an object, whereas mice with a mutation in SHANK2 (top) split their time between the two.
Two new strains of mice carrying different mutations in the SHANK2 gene show similar autism-like behaviors but opposing effects on brain signaling, according to two independent studies published 14 June in Nature1,2.
SHANK2 encodes a protein that supports synapses, the junctions between neurons. Deletions in the gene have been identified in a handful of individuals who have autism or intellectual disability. Several other synaptic proteins, such as the related SHANK3 and neuroligins and neurexins, have also been linked to autism.
One of the strains, developed by Korean researchers, has lower-than-normal activity at so-called NMDA receptors, triggered by the neurotransmitter glutamate. Drugs that boost NMDA signaling restore these animals’ social behaviors but not their repetitive behaviors or hyperactivity, the researchers found.
The second strain, first published online in April by Tobias Boeckers and various collaborators across Europe, shows an increase in signaling at NMDA receptors.
The findings highlight how different mutations in the same gene can lead to profoundly different molecular effects, making it challenging for drug makers to develop treatments that will be effective across the diverse autism spectrum.
“The mice are very opposite in some measures of synaptic function, and that’s very interesting,” says Ozlem Bozdagi-Gunal, assistant professor of psychiatry at Mount Sinai School of Medicine in New York City, who was not involved in either study.
It will take a long time, Bozdagi-Gunal adds, to understand how these molecular differences lead to the same behaviors in mice, and how they contribute to autism in people. Unlike mouse models of fragile X syndrome, which have relatively well-characterized molecular underpinnings, “in SHANK models, the interactions are more complicated,” she says.
Reverse results:
The SHANK2 mouse developed by Boeckers debuted last year at the Society for Neuroscience annual meeting, and carries a mutation in exon 7. (An exon is one piece of a protein-coding section of the genome.) In contrast, mice in the Korean study lack exons 6 and 7 — the same regions missing in individuals with autism.
Because both mutations interfere with the normal workings of the SHANK2 protein, it’s unclear why they cause opposite patterns in neuron signaling, notes Min Goo Lee, professor of pharmacology at Yonsei University College of Medicine in Seoul, who helped develop the Korean SHANK2 mouse.
Given other recent results, however, it’s not surprising that subtle differences in the type of mutations can have dramatically diverse effects in the brain, says Bozdagi-Gunal.
In the past couple of years, her team and several others have made mouse models carrying mutations in different parts of SHANK3. These models show a puzzling array of abnormalities. For example, one has severe social deficits and repetitive grooming, whereas another has more subtle social problems.
This may be because different domains of a protein have different functions. “The binding proteins for one domain are different than another domain, and so that can make the signaling different,” she says.
The implications for drug development are similarly complex.
In the new study, the researchers reversed many of the animals’ social deficits by giving one of two drugs. The first, called D-cycloserine, was originally used as an antibiotic for treating tuberculosis. Researchers later discovered that it partially activates NMDA receptors, leading to its testing for psychiatric disorders such as schizophrenia3 and obsessive-compulsive disorder4, with promising preliminary results.
The new findings support research published in 2010, which showed that the drug relieves repetitive grooming behaviors in mice lacking the autism-linked protein NLGN1, which also functions at synapses.
The drug’s effects in people are unclear, though a 2004 pilot study tested it in ten children and young adults with autism and found that it improves social interactions5. No follow-up studies have been published, however. The drug has several known side effects, including irritability, depression and convulsions.
The other drug tested in the new study is CDPPB, which indirectly boosts NMDA function by activating another glutamate receptor called mGluR5. Intriguingly, mGluR5 is overactive in mouse models of fragile X syndrome, and several clinical trials are testing mGluR5 blockers to treat fragile X syndrome and autism.
That both mGluR5 activators and blockers seem to alleviate symptoms of autism in different animal models “is particularly perplexing,” says Craig Powell, professor of neurology and neurotherapeutics at the University of Texas Southwestern in Dallas. He points out, though, that some forms of autism stem from either a deletion or a duplication of the same gene. Another study, published late last year, found opposite molecular signatures in mouse models of fragile X syndrome and tuberous sclerosis, both autism-related disorders.
“This may represent another ‘Goldilocks’ analogy where too much and too little of a good thing are deleterious,” Powell says. “In the treatment of autism, we should certainly proceed with caution and aim for ‘just right.’”
By joining the discussion, you agree to our privacy policy.