
THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A new strain of mice carries large mutations that more closely mimic the genetic variations seen in some people with autism than do two previously created strains. Together, the mouse models may help researchers understand autism and test new treatments for the condition.
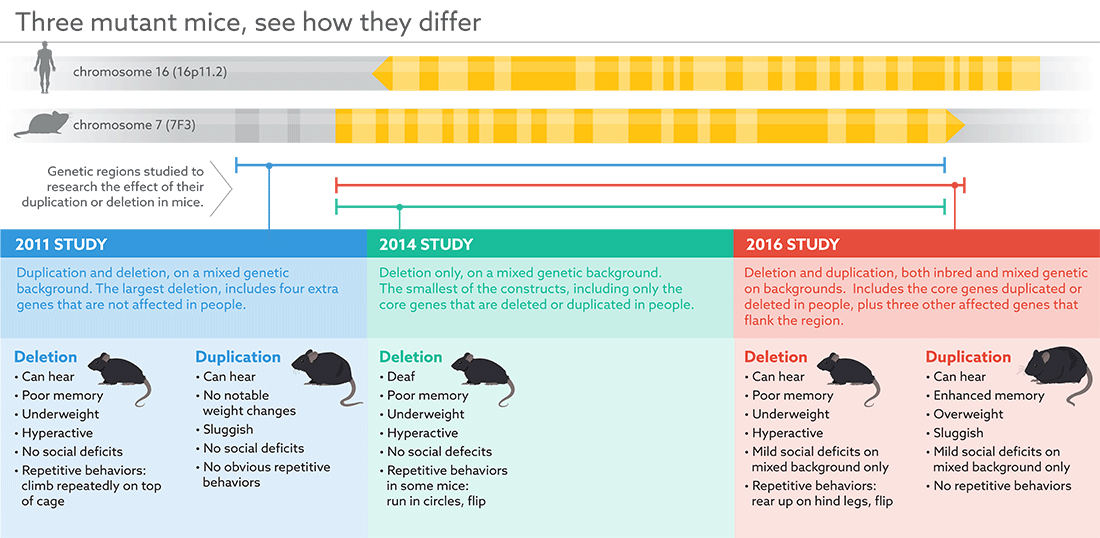
The mice either lack a copy of a region on chromosome 16 called 16p11.2, or carry an extra copy of that segment. Roughly 1 percent of people with autism have a duplication or deletion of the region, leading to one or three copies rather than the usual two.
In previous efforts to engineer mice with this so-called copy number variant (CNV), researchers deleted or duplicated slight variations of this region (see graphic below). The new mice may harbor the version most similar to the affected region in people.
“We created new mice, which we think are closer — at least genome-wise — to what’s happening in human[s],” says Alexandre Reymond, associate professor at the University de Lausanne in Switzerland. The work appeared 12 February in PLoS Genetics1.
Reymond’s team found that, like the previous strains of mice, the new mice show some of the key features — such as cognitive problems — associated with autism. “These mice are not fundamentally different,” Reymond says. Unlike the previous strains, however, some of the new mice show hints of the social deficits seen in people with autism.
The similarities between the mouse strains are striking, given the variability that plagues animal studies, says Jacqueline Crawley, professor of psychiatry and behavioral sciences at the University of California, Davis, who was not involved in the new work. Mice with the same mutation may behave differently, for example, if they differ in genetic background or simply hail from separate labs.
“Replication of cognitive deficits across four different labs working with three lines of independently generated 16p11.2 deletion mice is a truly remarkable accomplishment,” says Crawley, who has worked with one of the earlier strains.
Fine tuning:
Reymond and his colleagues engineered mice with either a deletion or a duplication of the core genes in 16p11.2, as well as three genes that bookend the region in people. The expression of these three genes is altered in people with the CNV, Reymond says, but possibly to a lesser extent than in the mice.
By contrast, the region in the first mouse model of 16p11.2, created in 2011, includes the core genes in human 16p11.2, two of the flanking genes and four other genes that are not affected in people2. The second mouse model, created in 2014, includes the core genes but no flanking genes3.
In all three models, the deletion mice are sick at birth, and many die shortly after.
Reymond’s team is the first to thoroughly compare features between mice with a deletion and those with a duplication. For instance, mice with a deletion are hyperactive and thin, whereas mice with a duplication tend to be sluggish. Only the mice with a deletion show repetitive behaviors reminiscent of those seen in people with autism.
In all three sets of models, mice with a deletion have cognitive deficits45. By contrast, the new study suggests, mice with a duplication have enhanced memory: They are more likely than controls to remember an object they have seen before. They are also overweight.
Like their rodent counterparts, people with the deletion tend to have lower intelligence quotients than their parents do, whereas those with the duplication may have an enhanced ability to remember words6.
The region also affects metabolism in people, but in the opposite direction from the mice: People with the deletion tend to be overweight and those with the duplication tend to be thinner than average. The reason for this metabolic difference between species is still a mystery, says Reymond.
Background effects:
There are other key differences among the three strains.
The mice created in 2014, which have a deletion of the region, are deaf. This strain carries a range of genetic variants, so the deafness may stem from interactions between genes in 16p11.2 and the other variants, the researchers say.
To better control for the effects of background genetic variation, Reymond’s team used inbred mice for their study, meaning that the the genetic background of these mice remains constant from one generation to the next.
To explore the effect of genetic background on the mice’s features, the researchers bred one generation of mice with another strain.
The cross-bred mice are similar to the inbred versions, but have healthier weights at birth. They also spend slightly less time sniffing and following cage-mates than controls do, hinting at social deficits. This is the first indication of a social deficit in a mouse model of the 16p11.2 mutations so far.
The contrast between the good health and social reticence of the cross-bred mice suggests that physical impairments and social deficits do not always go hand in hand, says Mu Yang, assistant professor of psychiatry and behavioral sciences at the University of California, Davis. Yang was not involved in the new work, but has worked with one of the earlier strains.
According to Reymond, the inbred strain is particularly useful because a large consortium of scientists is using the same background to engineer a mutant mouse for each of the nearly 20,000 genes in the mouse genome. Once this panoply of mice is available to researchers, they can compare mice missing each of the genes in the 16p11.2 region with mice lacking the whole region.
Lucy Reading-Ikkanda
By joining the discussion, you agree to our privacy policy.