THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
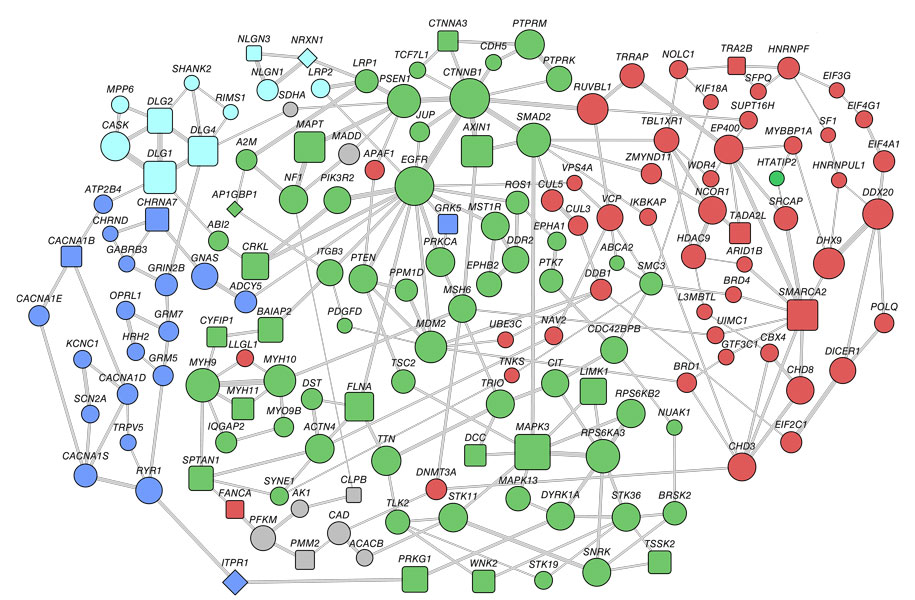
Moving beyond compiling long lists of genes that may be involved in autism, researchers are constructing networks: To find unifying threads among the embarrassment of genetic riches, they are stringing together autism genes to map connections between them — and, potentially, reveal clusters of biological insight.
Building the networks is an art in itself. Researchers must decide which genes should form the hub of the network and how to connect the genes — inherently subjective tasks that can deeply influence the results.
Two studies published in the past few months have tried to minimize researcher bias in the choice of genes1,2. They also connect the genes in creative ways. Their findings — that autism may stem from problems with motor skills and brain connections, for example — confirm results from other approaches, offering reassurance that researchers are on the right track.
“Genetics is a small part of what’s going on in autism,” says Michael Snyder, chair of genetics at Stanford University and lead investigator of one of the studies. “You really need to pin these pathways and networks to understand the biological factors that also play into it,” he says.
In the study, published 30 December in Molecular Systems Biology, his team began with a large database of gene and protein interactions called BioGrid1. The database is considered ‘naïve’ because it is not limited to autism genes but rather links more than 13,000 proteins based on their interactions.
The team looked for the clusters of interacting proteins — or ‘modules’ — that contain the largest numbers of autism candidates from a preset list of 383 genes.
“Two modules came out screaming.” says Snyder.
One module has genes that are mostly involved in neuronal signaling; the other has genes that control the expression of other genes by modifying DNA structure.
Brain connections:
Both functions are already strongly implicated in autism, but the study confirms their importance, says Stephan Sanders, assistant professor of psychiatry at the University of California, San Francisco, who was not involved in the work. “I think it’s very positive news that protein-protein interactions are hitting the same two broad categories of genes as other methods,” Sanders says.
The signaling cluster is also rich in genes that function mainly at the corpus callosum — a band of nerve fibers that connects the brain’s hemispheres. One-third of people who lack a corpus callosum meet diagnostic criteria for autism.
Snyder and his colleagues began with a large set of unrelated genes instead of autism candidates, which most network studies of this type use. Because of this, their study may avoid some selection bias, they say.
Still, the set of genes they applied to this network came from SFARI Gene, a database of autism-linked genes that in some ways is itself a biased set, says Dan Arking, associate professor of genetic medicine at Johns Hopkins University. (SFARI Gene is funded by the Simons Foundation, SFARI.org’s parent organization.)
The database is considered comprehensive and includes genes with only tangential links to autism. For example, some genes are listed mainly because they function at neuronal junctions, or synapses, which are thought to be pivotal in autism.
In the second study, published 22 December in Nature Neuroscience, Dennis Vitkup and his colleagues used a different method to try to understand autism’s diversity2.
They applied an algorithm called NETBAG, which connects genes that are likely to lead to similar symptoms when mutated. For example, the algorithm connects two genes if they share binding partners or are expressed at the same time during development.
The researchers used NETBAG to look for functional connections between 991 genes known to be spontaneously mutated in individuals with autism but not in their unaffected parents. Of these, 434 genes are part of large duplications or deletions called copy number variations.
The researchers found one tight cluster of 159 genes. They then used these genes to look for patterns that might underlie autism’s diversity. They found, for example, that spontaneous mutations are rare in people who have mild forms of autism.
They also found that genes mutated in girls with autism are expressed at higher levels than genes mutated in boys with the disorder. This finding supports the theory that girls require a bigger genetic hit to develop autism than boys do.
And the researchers found that the genes in the network function in most cell types, and throughout development, suggesting that looking for one origin for autism in the brain is misguided.
“I think that the belief that there will be ultimately a single brain area, or a single neuron type, where everything will converge is actually incorrect,” says Vitkup, associate professor of computational biology at Columbia University.
Still, this type of analysis can reveal areas that are important. “This gives a hint of what are the core issues,” he says. For example, the study found a key role for genes expressed highly in the striatum, a brain structure involved in planning movements.
In this study, the researchers used genes that had been statistically linked to autism in sequencing studies. This approach carries less bias than relying on a preselected list of autism candidates, Vitkup says.
“We didn’t start with a hypothesis of what causes autism,” he says. “We looked at the genome-wide data, which is unbiased, and we let the data tell us what [pathway] was the most affected.”
The two new studies use entirely different approaches but came to similar conclusions — which is an encouraging sign, Sanders says. Still, he says, it’s clear that network analyses will go only so far.
“We’re seeing everything converging on similar pathways, and I think that’s really cause for celebration,” Sanders says. “But we now know that just finding the genes is probably not going to be enough; we need to put them in the context of biology.”
References:
1: Li J. et al. Mol. Syst. Biol. 10, 774 (2014) PubMed
2: Chang J. et al. Nat. Neurosci. 18, 191-198 (2015) PubMed
By joining the discussion, you agree to our privacy policy.