THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
A modified version of the genetic engineering tool CRISPR restores expression of the gene mutated in fragile X syndrome, a new stem-cell study suggests1.
In particular, the tweak enables researchers to remove certain chemical tags, or methyl groups, on the gene that shut down production of a key protein.
The method could be used to treat people with fragile X syndrome, which leads to intellectual disability, social anxiety and, often, autism.
Fragile X syndrome stems from repeats of the DNA triplet CGG at the start of the FMR1 gene, which encodes a protein called FMRP. Typical individuals have fewer than 50 of these repeats, but people with fragile X have in excess of 200. The expansion adds methyl groups to the gene that block FMRP production.
CRISPR usually combines a molecular scissors with a DNA-binding protein that guides the scissors to specific stretches of DNA. In the new study, researchers replaced the scissors with an enzyme that removes the methyl groups. They used a guide RNA to target the repeats in FMR1.
They found that removing the tags from the repeats restores the gene’s expression in cells from people with fragile X.
The findings show conclusively that reversing methylation is the key to restoring FMR1 function, says lead researcher Rudolf Jaenisch, professor of biology at the Massachusetts Institute of Technology. “The question was: If you target the repeat, will it be sufficient to turn on the gene?” he says. The answer is yes.
Restoring a gene’s function is a better approach to treating genetic conditions than is patching up the effects of a mutation, says Arthur Beaudet, professor of molecular and human genetics at Baylor College of Medicine in Houston, Texas, who was not involved in the study. “This publication is an outstanding step in the right direction,” he says.
Long-term treatment:
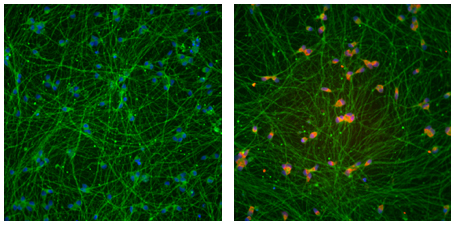
Jaenisch’s team started with a line of stem cells derived from the skin cells of an individual with fragile X syndrome. The FMR1 gene in this individual has 450 CGG repeats.
The modified CRISPR system removed the tags on the repeats, and this activated a region near the gene called a promoter, which controls the gene’s expression. The tool restored FMR1 expression to 90 percent of normal levels, yielding 73 percent of the typical amount of FMRP.
The effects persisted for at least two weeks, suggesting the treatment could lead to long-term benefits.
The technique works to a lesser degree on neurons derived from stem cells: It restored FMR1 expression to roughly half of typical levels. (The researchers did not quantify protein levels.)
The researchers also coaxed the modified stem cells to develop into precursors of neurons and introduced them into the brains of newborn mice. Roughly half of these cells, which became neurons, continued to express FMR1 three months later.
The possibility of using the new technique as a treatment in people with fragile X syndrome is exciting, says Randi Hagerman, medical director of the MIND Institute at the University of California, Davis, who was not involved in the study.
It isn’t possible to test the treatment in mice, as CGG repeats do not silence mouse FMR1. Prior to testing it in people, researchers would need to find a way to introduce the tool into each neuron, Hagerman says.
Off–target binding:
It is also important to ensure that the method does not lead to overexpression of FMR1, which can be toxic, Hagerman says. For example, individuals with 50 to 200 CGG repeats may develop a neurodegenerative syndrome that results from the gene making too much messenger RNA, she says.
The CRISPR enzyme binds to more than 1,000 other sites in the genome that contain CGG repeats, but has little effect on other genes in the stem cells. In neurons, it increases the expression of one gene ninefold and of 11 others up to fourfold — compared with the nearly 500-fold rise in FMR1 expression.
Jaenisch and his team hope to make the tool even more specific. They are also exploring whether the treatment is effective only at certain ages or stages of development.
The approach might also have promise for treating Rett syndrome, says Antonio Bedalov, associate professor of medicine at the University of Washington in Seattle, who was not involved in the new work. Rett syndrome is related to autism and stems from mutations in the MECP2 gene on the X chromosome. Methylation randomly silences one of the two X chromosomes in girls with the condition.
‘Unsilencing’ the other X chromosome could turn on the intact version of MECP2 and ease Rett features, Bedalov says. The new method might enable researchers to target MECP2 without affecting other X chromosome genes.
By joining the discussion, you agree to our privacy policy.