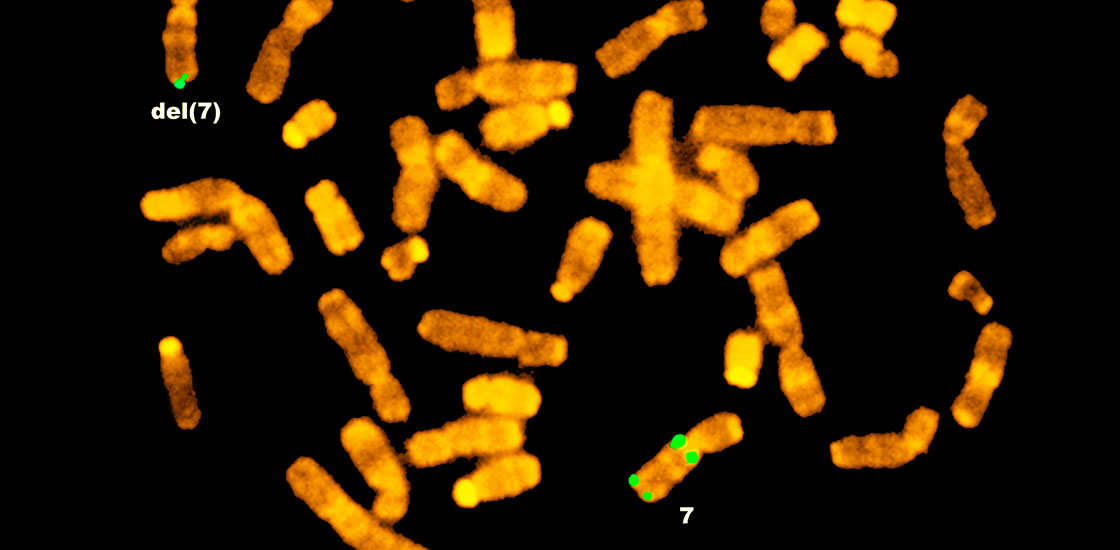
Having one too few or too many copies of a region on chromosome 7 leads to widespread changes in the chemical marks that direct gene expression, according to a study published 6 August in The American Journal of Human Genetics1. Many of these marks fall within autism genes, suggesting that these changes may contribute to the disorder.
The gain or loss of DNA within this mutational hotspot on chromosome 7, called 7q11.23, leads to two different syndromes, both of which share features with autism. Deletions in the region cause Williams syndrome, a condition characterized by developmental delays, anxiety and medical complications such as heart and kidney problems.
Children with a duplication of the region have what is called Dup7 syndrome. They experience a similar increase in anxiety, and show repetitive behaviors as well as social deficits.
The 7q11.23 region contains 25 genes, 6 of which have ‘epigenetic’ effects — meaning they modify gene expression without altering DNA sequence.
The researchers investigated how alterations of 7q11.23 affect one type of epigenetic change, the addition of methyl groups to DNA, which typically blocks a gene’s expression. They took blood samples from 20 children with Williams syndrome, 10 with Dup7 syndrome and 15 controls.
Their search turned up 1,413 locations at which children with Williams syndrome and their typically developing peers have different numbers of methyl groups. They found 508 such discrepancies between unaffected children and those with Dup7 syndrome.
Comparing children who have deletions with those who have duplications in 7q11.23, however, revealed a whopping 5,453 sites across the genome with methylation differences.
“[The findings] show how changes in a relatively small number of genes can have such dramatic and wide-ranging effects,” says lead researcher Lucy Osborne, professor of medical genetics at the University of Toronto.
For more than 2,000 genes, a deletion in 7q11.23 leads to a change in methylation that is opposite to that of the duplication. In people with the deletion, these genes usually sport more methyl groups than they do in Dup7 cells. (The list of 2,000 includes 47 genes associated with autism, including SHANK2, DYRK1A and CNTNAP2.)
This observation mirrors the contrasting social behavior of children with Dup7 syndrome, who tend to be socially withdrawn, and those with Williams syndrome, who are unusually gregarious.
The parallels between the genetic and behavioral observations are intriguing, says Hongjun Song, professor of neurology at Johns Hopkins School of Medicine in Baltimore, who was not involved with the study. Song notes, however, that it is still not clear how changes to the genes in the 7q11.23 region lead to alterations in methyl groups and, ultimately, symptoms.
It will also be important to check whether the same changes are seen in neurons, which have different epigenetic patterns than blood cells do, he notes. In fact, many of the genes with altered methylation are known to play an important role in the developing brain and are not even produced in blood cells.
That these epigenetic changes show up in blood cells suggests that they occur early in development, before all of the body’s cells have become specialized. “Probably whatever is going awry in establishing the methylation pattern is happening very early, which is why we see that pattern, even if it is not relevant to that tissue,” Osborne says.
Examining neurons directly is the next key step. Osborne and her colleagues are taking skin cells from children with Williams syndrome or Dup7 and coaxing the cells to become neurons in a dish. The researchers can then examine them to determine if their mixed-up methylation patterns alter gene expression and, ultimately, symptoms.