We have learned over the years that the genetic landscape of autism is complex. The enormous diversity of RNA and protein modifications will certainly add to that complexity, as will environmental influences. But as the notable papers from 2012 show, we are, at long last, on the way.
We have learned over the years that the genetic landscape of autism is complex. The enormous diversity of RNA and protein modifications will certainly add to that complexity, as will environmental influences. But as the notable papers from 2012 show, we are, at long last, on the way.
Several papers this year clarified the genetic landscape of autism. The four papers I discuss below all searched for highly penetrant de novo, or spontaneous, mutations in simplex families.
Three of the four studies are based on families in the Simons Simplex Collection (SSC), funded by the Simons Foundation, SFARI.org’s parent organization. The quad structure — with samples from two unaffected parents, one child with autism and an unaffected sibling — added statistical power to the findings.
Although they are individually rare, current estimates are that more than 400 ‘killer’ nonsense mutations will be found, and may account for a large fraction of all idiopathic cases of autism — those with an unknown cause.
A key criterion is recurrence. The studies have identified a number of variants in multiple individuals, and the number will surely rise as researchers complete the SSC analyses and screen other cohorts for likely candidates.
The first analyses of gene and protein networks point strongly to structural proteins at excitatory and inhibitory connections between neurons, to the immune system, and to chromatin structure.
About 30 percent of individuals with fragile X syndrome have traits of autism. The discovery that there is a significant overlap between de novo autism risk factors and a subset of proteins that interact with FMRP — the protein missing in fragile X syndrome — is of great importance.
Common effect:
Rare de novo variants are not the only source of genetic variation that enhances the risk of autism.
In their paper, Klei et al. provided evidence that common variants that have small or negligible effect on their own may contribute to autism in 40 percent of simplex cases and more than 60 percent of multiplex families — those that have more one child with autism.
The emphasis on genetics certainly does not rule out roles for environmental influences.
Several observations point to an altered immune response, for example. Animal models of autism and postmortem brains of children and adults diagnosed with the disorder have been shown to have more activated astrocytes and microglia than controls do. Studies have also found altered cytokine levels in postmortem brains, cerebrospinal fluid and plasma from people with autism.
Two papers this year addressed the issue of cause and effect using bone marrow transplantation in a mouse model of Rett syndrome and an immune model of autism.
In the Rett mice, the transplant improves respiratory and other symptoms, and normalizes various markers of the immune response. The treated mice also become more active and live longer than untreated mice. But how microglia — which the researchers reported as being crucial in the treatment — do their job remains unclear.
In the area of therapeutics, drugs that mimic the action of gamma-aminobutyric acid, or GABA, the neurotransmitter that mediates inhibitory signaling, held center stage.
It has become accepted that one correlate of autism is an imbalance between excitation and inhibition (E/I) in critical regions of the brain. Although one cannot be more precise at present, attempts are under way to manipulate this balance with drugs that enhance signaling through GABA and dampen signaling through the excitatory neurotransmitter glutamate.
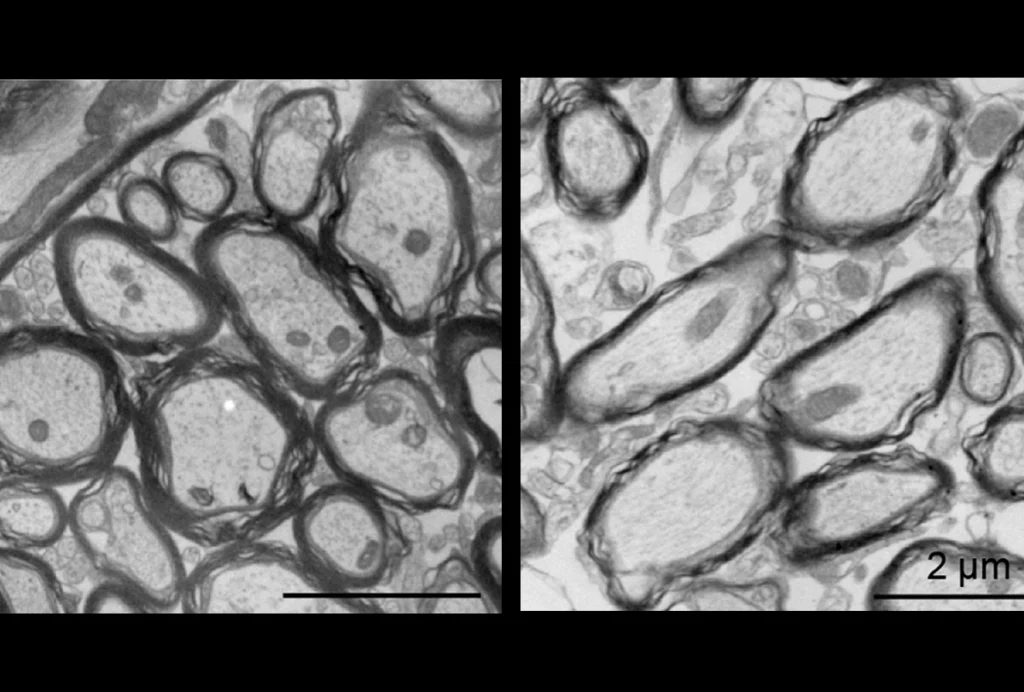
Arbaclofen, a GABA-B agonist, proved somewhat effective in two trials of fragile X syndrome. In the first, a double-blind, placebo-controlled crossover study of 65 individuals with the full fragile X mutation, the researchers observed “positive trends” in “multiple global measures.”
Another group studied arbaclofen in mice lacking FMR1, the fragile X gene. The researchers found that arbaclofen restores protein synthesis in the mouse brains to normal levels. Most significantly, chronic administration of arbaclofen decreases the density of dendritic spines — the signal-receiving branches of neurons — in the mutant mice, but not in controls.
Brain dysfunction:
In another elegant paper, researchers described how eliminating a gene called TAOK2 changes the shape of dendrites. This gene is present in the chromosomal region 16p11.2, which is strongly linked to autism. TAOK2 interacts with three signaling proteins, NRP1, SEMA3A and JNK. These interactions may be a major factor in setting E/I balance.
Some of the first studies of mouse models and human autopsy material found evidence for altered cerebellar function in autism. A paper published this year demonstrated that the number of Purkinje cells drops by 60 percent after two months and 80 percent after four months in mouse models of tuberous sclerosis complex.
The mice also show drastic alterations in the shape of the hallmark Purkinje cell dendritic tree, with reduced spine density. They have several autism-like behaviors, including lack of interest in another mouse, repetitive grooming, and more vocalizations compared with controls. Interestingly, the loss in Purkinje cells occurs long after the altered behaviors are first observed. It is encouraging that all of these symptoms can be prevented and reversed by treatment with rapamycin.
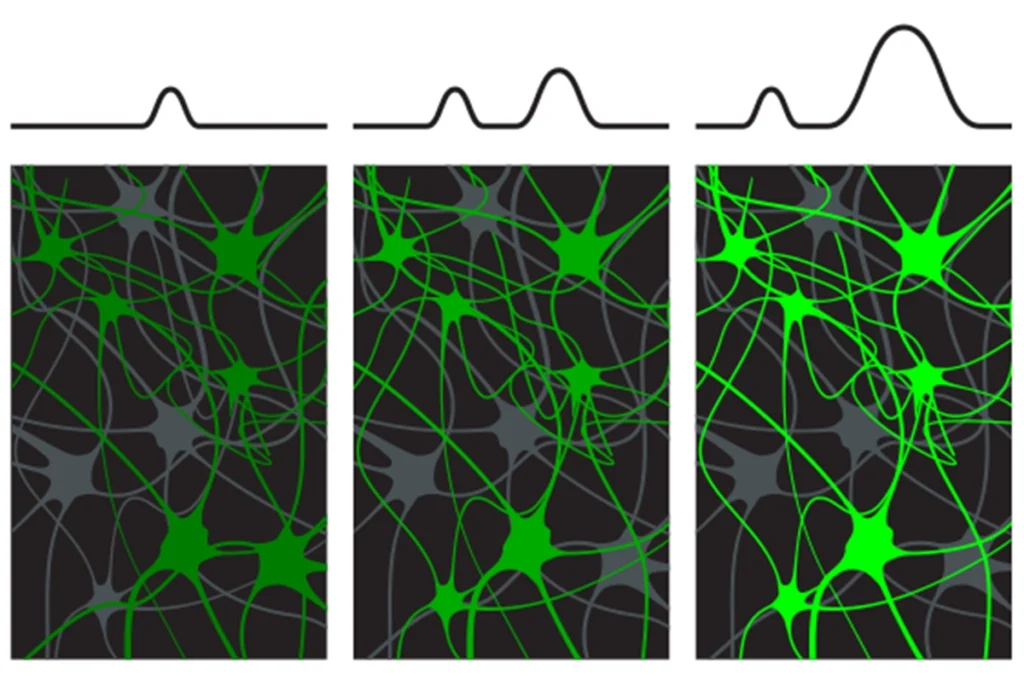
Despite the marked heterogeneity of autism in the human population and in animal models, most investigators focus on mean responses. The range of responses is large, so many animals are needed. In particular, electrophysiological or functional magnetic resonance imaging recordings from the cerebral cortex are particularly variable.
One important paper called attention to the possibility that the variation between cortical responses may contain significant information. By examining several sensory modalities, the researchers found that the mean responses are no different in high-functioning individuals with autism and neurotypical controls, but the variability of responses is different between the two groups. They ruled out motion artifacts with clever experiments using global responses recorded in the same regions of interest.
This introduces a new, unexpected concept of an inherently ‘noisy brain’ in people with autism that must be explained and might possibly serve as a biomarker for the disorder. Assays of gene function in mouse models should perhaps examine individual variability as well as changes in mean response.
The challenge for 2013 is to bring these findings in genetics, neural circuits and behaviors one step closer together, and to use all of the information to think of new approaches to therapeutics.